Fenilcetonúria - sinais clássicos de transmissão hereditária e dietoterapia
Conteúdo
- 1Como se manifesta a fenilcetonúria
- 2Mecanismo de desenvolvimento de doenças
- 3Fenilcetonúria em crianças
- 4Sintomas da doença
- 5Causas e gatilhos
- 6Diagnóstico
- 7Tratamento da fenilcetonúria clássica
- 8Características da nutrição de recém-nascidos edietoterapia
- 9Dieta para crianças em idade pré-escolar e escolares
- 10Grupos de produtos com PKU
- 11Como controlar o nível de fenilalanina no sangue
- )12Vídeo
As doenças cuja ocorrência está relacionada a defeitos no aparelho celular genético - fenilcetonúria - estão incluídas em uma pequena lista de doenças hereditárias tratáveis.O pioneiro dessa doença foi um médico norueguês IA Felling, mais tarde foi descoberto que um único gene chamado gene da fenilalanina hidroxilase (o braço longo do 12º cromossomo contendo até 4,5% de todo o material celular de DNA) foi responsável pelo desenvolvimento e curso da doença.Defeito hereditário leva à desativação parcial ou completa da enzima hepática fenilalanina-4-hidroxilase.
Como a doença por fenilcetonúria é detectada
A doença por fenilcetonúria hereditária (PKU) leva ao envenenamento crônico do corpo com substâncias tóxicas formadas como resultado do metabolismo e processo de aminoácidos prejudicadoshidroxilação de fenilalanina.A intoxicação permanente causa danos ao sistema nervoso central (SNC), cuja manifestação é um declínio progressivo da inteligência (oligofrenia fenilpirúvica).
A doença de Felling se manifesta no acúmulo excessivo no corpo de fenilalanina e nos produtos de seu metabolismo incorreto.Outros fatores no desenvolvimento da fenilcetonúria incluem transporte prejudicado de aminoácidos através da barreira hematoencefálica, baixa contagem de neurotransmissores (serotonina, histamina, dopamina).Na ausência de tratamento oportuno da doença, leva ao retardo mental e pode causar a morte da criança.

O mecanismo de desenvolvimento da doença
A causa dos distúrbios genéticos é um bloqueio metabólico que impede a formação de fenilalanina-4-hidroxilase (uma enzima,responsável pela conversão do aminoácido fenilalanina em tirosina).O aminoácido proteinogênico tirosina é parte integrante das proteínas e do pigmento da melanina, por isso é um elemento necessário para o funcionamento de todos os sistemas do corpo, e sua falta leva à fermentopatia.
A inibição da formação de metabólitos causada pela inativação mutacional da enzima é a ativação de vias metabólicas auxiliares da fenilalanina.O alfa-aminoácido aromático, como resultado de processos metabólicos defeituosos, se divide em derivados tóxicos, que em condições normais não se formam:
- ácido fenilpirúvico (fenilpiruvato) - um alfa-cetoácido gordo-aromático, sua formaçãoleva à mielinização de processos neuronais e demência;
- o ácido fenil láctico é um produto formado durante a recuperação do ácido fenilpirúvico;
- feniletilamina - o composto inicial para transmissores biologicamente ativos de impulsos eletroquímicos, aumenta a concentração de dopamina, adrenalina e norepinefrina;
- O acetato de ortofenil é uma substância tóxica que causa distúrbios metabólicos no cérebro.
As estatísticas médicas indicam que um gene patologicamente alterado está presente em 2% da população, mas não se manifesta de forma alguma.O defeito genético é transmitido à criança pelos pais apenas na presença da doença nos dois parceiros, com o bebê em 50% dos casos se tornando o portador do gene mutado, permanecendo saudável.A probabilidade de fenilcetonúria em recém-nascidos levar à doença é de 25%.
Por qual tipo é herdado
A doença de Felling é uma anormalidade genética herdada de um tipo autossômico recessivo.Esse tipo de herança significa que o desenvolvimento de sinais de uma doença congênita só ocorrerá quando uma cópia genética defeituosa de ambos os pais, portadores heterozigotos do gene alterado, for herdada da criança.
O desenvolvimento de uma doença congênita em 99% dos casos é causado por uma mutação no gene responsável pela codificação da enzima que fornece a síntese da fenilalanina-4-hidroxilase (fenilcetonúria clássica).Até 1% das doenças genéticas estão relacionadas a alterações mutacionais que ocorrem em outros genes que causaminsuficiência de dihidrteridina redutase (PKU tipo II) ou tetra-hidrobiopterina (PKU tipo III).
Fenilcetonúria em crianças
Na maioria dos casos, a forma clássica de doença genética em crianças manifesta-se externamente perceptível, a partir dos 3-9 meses de idade.Os recém-nascidos com um gene defeituoso, parecem saudáveis, uma característica distintiva é o hábito específico (aparência) do bebê.A sintomatologia expressa aparece durante 6-12 meses após o nascimento.
A PKU tipo II é caracterizada pelo fato de os primeiros sintomas clínicos aparecerem 1,5 anos após o nascimento.Os sinais da doença não desaparecem após o diagnóstico de anormalidades genéticas e o início da dietoterapia.Esse tipo de doença congênita geralmente leva a um resultado fatal por 2-3 anos da vida de uma criança.Os sintomas mais comuns da PKU tipo II são:
- transtornos mentais graves;
- hiperreflexia;
- função motora comprometida de todas as extremidades;
- síndrome de contrações musculares não controladas.
Os sinais clínicos de alterações mutacionais nos genes do tipo III são semelhantes à doença do tipo II.A deficiência de tetra-hidrobiopterina é caracterizada por uma tríade de sintomas específicos:
- um alto grau de retardo mental;
- o tamanho do crânio é claramente reduzido em relação a outras partes do corpo;
- espasticidade muscular (é possível a perda completa da mobilidade dos membros).

Manifestações da doença de Felling
Em ensaios clínicos e observações, sugeriu-se quederivados tóxicos do metabolismo da fenilalanina causam um declínio na capacidade intelectual, que é de natureza progressiva e pode levar à demência (oligofrenia, idiotice).Entre as causas prováveis de distúrbios irreversíveis da atividade cerebral das mais razoáveis, considera-se que é causada por uma diminuição no nível de falta de tirosina de neurotransmissores que transmitem impulsos entre os neurônios.
A relação exata de causa e efeito entre doença hereditária e distúrbios cerebrais ainda não foi identificada, assim como o mecanismo de desenvolvimento devido à fenilcetonúria de estados mentais como echopraxia, ecolalia, ataques violentos e irritabilidade.Os resultados da análise indicam que a fenilalanina tem um efeito tóxico direto no cérebro, o que também pode causar uma diminuição na inteligência.
Estrutura e características fenotípicas
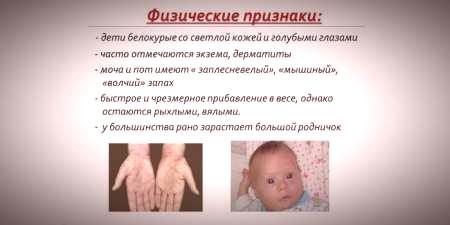
Dado que a saturação de pigmentos da pele e do cabelo depende do nível de tirosina nas mitocôndrias dos hepatócitos, e a fenilcetonúria leva a uma interrupção da conversão da fenilalanina, os pacientes com esta doença têm peculiaridades.sinais recessivos).O aumento do tônus muscular causa o aparecimento de desvios na estrutura do corpo - torna-se displásico.As características externas distintas da fenilcetonúria incluem:
- hipopigmentação - pele clara, olhos azuis pálidos, cabelos descoloridos;
- cianose das extremidades;
- tamanho reduzido da cabeça;
- posição específica do corpo - ao tentar ficar em pé ou sentada, a criança adota uma postura de "alfaiate" (braços e pernas dobrados nas articulações).
Sintomasdoença
Com a detecção oportuna, a doença de Felling é tratada com sucesso ajustando a nutrição e o desenvolvimento da criança ocorre de acordo com a faixa etária.A dificuldade de detectar uma mutação genética é que os sinais precoces são difíceis de detectar, mesmo por um pediatra experiente.A gravidade dos sintomas da doença congênita aumenta à medida que a criança cresce, pois o consumo de alimentos protéicos contribui para o desenvolvimento de distúrbios do SNC.
Sinais em recém-nascidos
Durante os primeiros dias de vida de uma criança, é difícil detectar sinais de anormalidades anormais - o bebê se comporta naturalmente, não há atrasos no desenvolvimento.Os sintomas da doença começam a aparecer pela primeira vez 2-6 meses após o nascimento.Os pais devem estar atentos ao comportamento do bebê, caracterizado por baixa atividade, letargia ou, inversamente, ansiedade, hiper-excitabilidade.
Com o início da amamentação, os recém-nascidos com leite começam a entrar no corpo do recém-nascido com leite, que é um catalisador para o aparecimento dos primeiros sinais, indicando claramente que a doença começou a progredir.Manifestações clínicas específicas da doença incluem:
- vômito constante (geralmente considerado estreitamento congênito do goleiro);
- vômitos frequentes;
- sem resposta a estímulos externos;
- distonia muscular (tensão muscular reduzida);
- síndrome convulsiva (convulsões de natureza epiléptica ou não epiléptica).
Sintomas em crianças após 6 meses
Se a manifestação de uma doença genética não forocorreu (ou não foi observado) durante os primeiros 6 meses após o nascimento da criança e, após esse período, já é possível determinar com precisão o atraso no desenvolvimento psicomotor.Os sintomas de distúrbios genéticos causados por deficiência enzimática em crianças com mais de seis meses são:
- atividade reduzida (até completa indiferença);
- falta de autocontrole, assento;
- um odor peculiar de "rato" da pele (o odor de mofo resulta da excreção de derivados tóxicos da fenilalanina através das glândulas sudoríparas e na urina);
- perda de capacidade de reconhecer visualmente o rosto dos pais;
- descamação da pele;
- dermatite, eczema, esclerodermia.
Progressão da doença na ausência de tratamento na infância
Se anormalidades no desenvolvimento não foram detectadas na infância e o tratamento apropriado não foi realizado, entãoa doença começa a progredir ativamente e muitas vezes leva à incapacidade.A falta de terapia em um estágio inicial da doença faz com que os seguintes sintomas se desenvolvam aos 1,5 anos de idade:
- microcefalia (tamanho cerebral diminuído);
- prognatia (deslocamento da dentição superior para frente);
- dentição tardia;
- hipoplasia do esmalte (desbaste ou ausência completa de esmalte dentário);
- o atraso no desenvolvimento da linguagem até a completa ausência de fala;
- 3, 4 grau de oligofrenia (retardo mental, retardo mental);
- defeitos cardíacos congênitos (defeitos na estrutura do músculo cardíaco, partes do coração,embarcações de grande porte);
- distúrbios do sistema autonômico (acrocianose, sudorese, hipotensão arterial);
- constipação.
Causas e fatores provocadores
Para a manifestação de uma mutação autossômica recessiva, um gene com defeito deve ser herdado de ambos os pais.Doenças genéticas desse tipo ocorrem com a mesma frequência em meninos e meninas recém-nascidos.A patogênese da PKU é causada pelo metabolismo prejudicado da fenilalanina, que pode ocorrer em três formas.A terapia dietética é submetida apenas à fenilcetonúria clássica do tipo I.
Formas atípicas da doença podem ser curadas ajustando a nutrição.Esses desvios são causados por uma deficiência de tetra-hidropinina, desidropinina redutase (raramente - piruviltiltetra-hidropinina sintase, guanosina-5-trifosfato e outros).A maioria dos casos fatais é registrada entre pacientes com variações raras de PKU, com manifestações clínicas de todas as formas da doença semelhantes.O risco de ter um bebê com um gene mutado da fenilalanina hidroxilase aumenta se os pais forem parentes próximos (em casamentos intimamente relacionados).
Diagnóstico
No caso de suspeita de desordens genéticas, o diagnóstico é feito com base em um conjunto de dados obtidos no estudo da história médica - dados genealógicos, resultados de estudos genéticos clínicos e médicos.Para a detecção oportuna de doenças congênitas (PKU, fibrose cística, galactosemia, etc.), foi desenvolvido um programa de massa compulsóriaexame laboratorial de todos os recém-nascidos (triagem neonatal).
Se os futuros pais estão cientes do portador do gene mutado, a medicina moderna oferece maneiras de detectar um defeito no estágio da gravidez (diagnóstico pré-natal do feto pelo método invasivo).Para a divisão da fenilcetonúria em tipos por gravidade, é utilizada uma classificação condicional baseada no nível de fenilalanina em um fluido plasmático do sangue livre de fibrinogênio:
- Fenilcetonúria pesada - 1200 μmol /l.
- Média - 60-1200 µmol /l.
- Leve (sem necessidade de tratamento) - 480 μmol /L.
Teste de triagem
A detecção de anormalidades genéticas ocorre em vários estágios.Na primeira etapa da maternidade, todos os bebês por 3-5 dias de vida recebem amostras de sangue periférico (de cinco) para pesquisa.O material é aplicado em papel e enviado ao laboratório bioquímico, onde é realizada a análise bioquímica.Na segunda etapa do teste de triagem, é determinada a concentração da concentração normal de fenilalanina.

Se nenhuma alteração patológica for detectada, o diagnóstico será concluído e o cartão da criança será registrado.No caso de desvios da norma, os resultados do diagnóstico são enviados ao pediatra para garantir um exame mais preciso da amostra de sangue do recém-nascido.A saúde da criança depende da implementação oportuna e precisa de todas as medidas para detectar desvios.Se o diagnóstico for confirmado após triagem repetida, os pais da criançaenviado a uma clínica de genética pediátrica para tratamento.
Análises e estudos para confirmar o diagnóstico
O re-diagnóstico após a detecção durante o teste de triagem primário de anormalidades é realizado reenviando os testes.Além de determinar o conteúdo de fenilalanina no sangue para os métodos de diagnóstico de PKU em crianças e adultos, incluem:
- Teste de queda - determinação de ácido fenilpirúvico na urina pela adição de cloreto de ferro ao biomaterial (coloração em azul esverdeado);
- Teste de Guthrie - avaliação do grau de resposta dos microrganismos ao metabolismo ou enzimas contidas no sangue do paciente;
- cromatografia - o estudo das propriedades químicas de substâncias distribuídas entre duas fases;
- fluorimetria - irradiação de biomaterial por radiação monocromática para determinar a concentração de substâncias nele contidas;
- eletroencefalografia - diagnóstico de atividade elétrica do cérebro;
- A ressonância magnética é a excitação dos núcleos atômicos das células por ondas eletromagnéticas e a medição de sua resposta.
Tratamento da fenilcetonúria clássica
No coração da fenilcetonúria, a terapia é uma restrição ao consumo de produtos que são a fonte de proteínas animais e vegetais.O único método de tratamento bem-sucedido é a dietoterapia, cuja adequação é avaliada pelo conteúdo de fenilalanina no soro.Nível máximo de aminoácidos em pacientes de diferentes faixas etáriasé:
- em bebês e crianças de até 3 anos - até 242 µmol /l;
- em pré-escolares de até 360 µmol /l;
- em pacientes de 7 a 14 anos - até 480 μmol /l;
- em adolescentes - até 600 μmol /l.
A eficácia da dieta depende de qual estágio da doença a dieta é corrigida.No diagnóstico precoce da patologia congênita, a dietoterapia é prescrita a partir da 8ª semana de vida (após esse período, alterações irreversíveis já começam).Ausência de medidas oportunas leva a complicações e uma diminuição do nível de inteligência em 4 pontos por 1 mês desde o nascimento até o início do tratamento.

Considerando que a dieta terapêutica da fenilcetonúria fornece exclusão completa da dieta das proteínas animais, é necessário o uso de outras fontes de aminoácidos essenciais, além de vitaminas do grupo B, cálcio -e compostos minerais contendo fósforo.Os produtos a serem usados como suplementos sem proteína incluem:
- hidrolisados de proteína (Amigen, Aminazole, Fibrinosol);
- não contêm misturas de fenilalanina saturadas com aminoácidos essenciais - Tetrafen, livre de fenil.
Além de medidas curativas para eliminar a causa do comprometimento do funcionamento do corpo, um tratamento sintomático deve ser realizado com o objetivo de eliminar defeitos de fala e normalizar a coordenação dos movimentos.A terapia complexa inclui procedimentos de fisioterapia, massagem, a ajuda de um fonoaudiólogo, um psicólogo, realizando exercícios de ginástica.Em alguns casos, em conjunto com dietoterapiamostra o uso de anticonvulsivantes, drogas nootrópicas e vasculares.
Peculiaridades do tratamento de formas atípicas
A fenilcetonúria tipo II e III não é tratável com dieta pobre em proteínas - o nível de fenilalanina no sangue permanece inalterado enquanto limita o fluxo de proteína no corpo ou os sintomas clínicos progridem mesmo com a diminuição do nível de aminoácidos.A terapia eficaz dessas formas da doença é realizada com o uso de:
- tetra-hidrobiopterina - fator da enzima afetada;
- análogos sintéticos da tetra-hidrobiopterina - essas substâncias penetram melhor na barreira hematoencefálica;
- medicamentos para terapia de substituição - não eliminam a causa da fenilcetonúria, mas apóiam o funcionamento normal do corpo (Levodopa juntamente com Carbidofa, 5-oxitriptofano, 5-formiltetra-hidrofolato);
- hepatoprotetores - suportam a função hepática;
- anticonvulsivantes;
- a introdução do gene da fenilalanina hidroxilase no fígado é um método experimental.
Nutrição para recém-nascidos e dietoterapia
A amamentação é permitida no primeiro ano de vida de um bebê com PKU, mas deve ser restrita.Até 6 meses, a ingestão aceitável de fenilalanina é de 60 a 90 mg por 1 kg de peso do bebê (100 g de leite contém 5,6 mg de fenilalanina).A partir de 3 meses, a dieta do bebê deve ser gradualmente expandida, introduzindo sucos e purês de frutas.
Crianças de 6 meses de idade podem fazer dieta de purê de legumes, mingau (sagu), sem proteínaácido.Após 7 meses, é possível dar ao bebê massas com poucas proteínas, a partir de 8 meses - pão, que não contém proteínas.A idade em que a proteína no corpo da criança doente deve ser restringida não foi estabelecida.Os médicos ainda estão discutindo a viabilidade da terapia dietética por toda a vida, mas concordam que uma dieta de pelo menos 18 anos deve ser seguida.
A fenilcetonúria diagnosticada em uma mulher não é um motivo para recusar o nascimento de um filho.Gestantes com PKU para prevenir lesões fetais durante a gravidez e para evitar possíveis complicações, é necessário observar uma dieta com dieta restrita a fenilalanina (no momento do parto) (até 242 μmol /l).
Misturas sem lactose para bebês
A dieta para fenilcetonúria é baseada em uma redução significativa na quantidade de proteína natural na dieta diária, mas o corpo da criança não pode se desenvolver normalmente na ausência dos oligoelementos necessários.Para atender à necessidade de proteína do bebê, são usadas misturas de aminoácidos sem lactose, que, segundo a lei russa, devem ser fornecidas gratuitamente.
A tolerância da criança à fenilalanina muda rapidamente durante o primeiro ano de vida, portanto sua concentração no sangue do bebê deve ser monitorada e a dieta ajustada.As misturas são projetadas para determinadas faixas etárias:
- bebês com menos de um ano são prescritos Afenilak 15, Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- Crianças com mais de 1 anoano, nomear enriquecido com misturas de vitaminas e minerais com alto teor de proteínas - PKU Prima, P-AM Universal, PKU-1, PKU-2, Maximeid XP, XP máximo.

Alimentos para suplementos de proteína
Um dos principais componentes de uma dieta para a fenilcetonúria são os produtos à base de amido com baixa proteína.Esses suplementos contêm hidrolisado de caseína, triptofano, tirosina, metionina, nitrogênio e fornecem as necessidades diárias do bebê para a proteína necessária para o desenvolvimento e crescimento normais.Os alimentos especializados que preenchem a falta de minerais e aminoácidos essenciais quando estão ausentes na dieta são:
- Berlofen;
- Zimorgan;
- Minafen;
- Aponti.
Dieta para crianças em idade pré-escolar e escolar
À medida que o corpo se ajusta à fenilalanina, crianças a partir dos 5 anos de idade podem reduzir gradualmente suas restrições alimentares.A expansão de uma dieta ocorre pela introdução de cereais, laticínios, produtos à base de carne.Os alunos mais velhos já têm uma alta tolerância à fenilalanina; portanto, nessa idade, você pode continuar a expandir a dieta, enquanto monitora a resposta a qualquer alteração na nutrição.Os seguintes métodos são usados para controlar a condição da criança:
- avaliação de parâmetros neurológicos, estado psicológico;
- controle do desempenho do eletroencefalograma;
- determinação do nível de fenilalanina.
Grupos de alimentos com PKU
A dieta de pacientes com PKU, juntamente com baixa proteínaprodutos amiláceos e misturas medicinais também incluem produtos de origem natural.Ao elaborar o menu, a quantidade de proteína consumida deve ser claramente calculada e a dosagem recomendada pelo médico não deve exceder.Para eliminar os efeitos tóxicos no corpo, foram desenvolvidas 3 listas de produtos que contêm itens proibidos (vermelho), não recomendados (laranja) e permitidos (verde).
Lista Vermelha
A fenilcetonúria se desenvolve no fundo da ausência de uma enzima que se converte em tirosina fenilalanina, de modo que um alto teor de proteínas é a base para listar os produtos na lista proibida (vermelha).Os itens desta lista devem excluir completamente a dieta de um paciente com PKU:
- carne;
- órgãos internos de animais, subprodutos;
- salsichas, salsichas;
- frutos do mar (incluindo peixes);
- ovos de todas as aves;
- produtos lácteos;
- porcas;
- leguminosas e cereais;
- produtos de soja;
- pratos contendo gelatina;
- confeitaria;
- aspartame.
Lista laranja
Os produtos a serem dosados para uma criança diagnosticada com PKU estão incluídos na lista laranja.A inclusão na dieta dos itens desta lista é aceitável, mas em um número estritamente limitado.Embora esses produtos não contenham muita proteína, eles também podem aumentar o nível de fenilalanina; portanto, seu uso não é recomendado:
- vegetais enlatados;
- pratos de batata e arroz;
- repolho;
- leite;
- sorvete.
Lista Verde
Produtos isentos de proteínas podem ser utilizados em pacientes com diagnóstico de fenilcetonúria sem restrição.Antes de comprar os itens da lista verde, é necessário examinar a composição indicada na embalagem e garantir que não haja corante para o aspartame contendo fenilalanina:
- frutas;
- vegetais (exceto batatas e couves);
- bagas;
- verdes;
- cereais amiláceos (sagu);
- mel, açúcar, geléia;
- produtos de farinha feitos de farinha de milho ou arroz;
- óleos, gorduras (manteiga, girassol, azeitona).
Como controlar o nível de fenilalanina no sangue
A fenilcetonúria é uma doença incurável que pode ser transferida para a fase de estagnação por meio de dieta e medidas terapêuticas.Na modificação de condições de vida, a violação de uma dieta a doença pode agravar-se novamente, por isso os pacientes precisam de observação ao longo da vida.O processo de controle é determinar periodicamente o nível de fenilalanina no sangue.A frequência do teste depende da idade do paciente:
- até 3 meses - a triagem de sangue deve ser realizada semanalmente para obter resultados consistentes;
- de 3 meses a 1 ano - 1-2 vezes por mês;
- 1 a 3 anos - uma vez a cada 2 meses;
- com mais de 3 anos - trimestralmente.

O sangue para análise é administrado 3-4 horas após a ingestão.Além da triagem, o desenvolvimento da PKU é controlado pela determinação do estado nutricional, do desenvolvimento físico e emocional do paciente, do nível das habilidades intelectuais.e desenvolvimento da linguagem.As observações podem exigir diagnósticos adicionais com o envolvimento de especialistas apropriados.
Vídeo
As informações apresentadas neste artigo são apenas para orientação.O artigo não pede auto-tratamento.Somente um médico qualificado pode diagnosticar e recomendar tratamento com base nas características individuais do paciente em particular.












